

LDA+DMFT method to X-ray spectroscopy
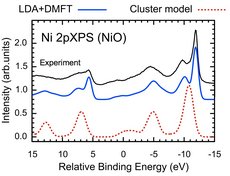
We have developed a package for 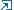 calculation of (core-level) X-ray absorption and photoemission spectra within LDA+DMFT. The core of the method is a quantum impurity solver based on truncated configuration interaction method. Compared to the conventional cluster-model approach, The present method is (almost) parameter free and can access fine spectral features in recent high-energy resolution experiments. We have used this method to study the non-local screening effects in series of transition metal oxides.
calculation of (core-level) X-ray absorption and photoemission spectra within LDA+DMFT. The core of the method is a quantum impurity solver based on truncated configuration interaction method. Compared to the conventional cluster-model approach, The present method is (almost) parameter free and can access fine spectral features in recent high-energy resolution experiments. We have used this method to study the non-local screening effects in series of transition metal oxides.
Linear response
We have developed a package for linear response (suscpetibilities) calculations within DMFT. This represents a non-trivial post-processing step, which requires calculation of two-particle (four-point) correlation functions and solution of a pair of Bethe-Salpeter equations. Our main goal is to calculate static suseptibilities and analyze their divergencies, in order to allow for an unbiased search for ordering instabilities. Our first non-trivial application of the approach was investigation of the two-band Hubbard model with the instability towards  excitonic condensation.
excitonic condensation.
Dynamical mean-field theory
Our main approach to interacting electron problems is the dynamical mean-field theory (DMFT). We use our own implementation of the self-consistency cycle, which allows treatment of multiple (inequivalent) impurities. Our main impurity solver is hybridization-expansion continuous-time Quantum Monte-Carlo (CT-HYB). We use several implementation of the algorithm: ALPS, w2dynamics and our own. We also use exact diagonalization solver. We have developed or are developing extentions of these solvers to allow treatment of off-diagonal (and complex off-diagonal hybridzation).
Density functional methods
Density functional theory allows to effectively reduce a many-body problem to the a one-particle problem. With this methodological simplification is it possible to study real materials, i.e. models that work in continuous space and use only positions of atomic nuclei as an input. In our work we use the Wien2k code based using LAPW basis set. In the past we have contributed to several packages of this code:
- LAPWSO (spin-orbit coupling)
- LAPWDM, ORB (LDA+U and other orbital potentials)
- QTL (projected densities of states)
- WIEN2WANNIER, WPLOT, WOPTIC (interface to wannier90 code)